Recently, new SARS-CoV-2 variants were detected in the United Kingdom and South Africa with multiple spike protein mutations that may cause substantial changes in certain properties of the virus. These observations spurred biosurveillance efforts in different countries, with some countries outside of the UK and South Africa already reporting the presence of these new variants within their territories. The data presented in this report is part of our own continuing biosurveillance efforts to track the entry of these new variants in the country, as well as other viral mutations that may be of concern locally.
In order to track if the new SARS-CoV-2 variants have already entered the country, in cooperation with the DOH Epidemiological Bureau, we have received samples from CAR, NCR, CALABARZON, and a single sample from General Santos and combined these with our samples biobanked at the PGC to sequence and analyze the genomes of a total of 305 SARS-CoV-2 samples that were collected from 31 October to 30 December 2020. These samples came from hospitalized patients as well as inbound travelers. All the samples included in this analysis had CT values below 30 in RT-PCR tests, suggesting moderate to high viral loads.
The samples were batch processed from 02 to 05 January, 2020 using the only next-generation sequencing workflow (https://www.illumina.com/products/by-type/ivd-products/covidseq.html) that has received Emergency Use Authorization (EUA) from both the US and the Philippine Food and Drug Administrations (FDA).
Among the 305 samples analyzed, we were able to assemble and assign lineages (Table 1) in 274 samples – none of which were classified to lineage B.1.1.7 (UK variant). We were also not able to observe the triple spike protein mutations characteristic of the South African variant (lineage B.1.351). The most common lineages observed in this set of samples are the following: B.1.1.63 (Hong Kong lineage) found in 106 samples, B.1.1 (European lineage) found in 51 samples, and B.1.1.263 (UAE lineage) found in 49 samples.
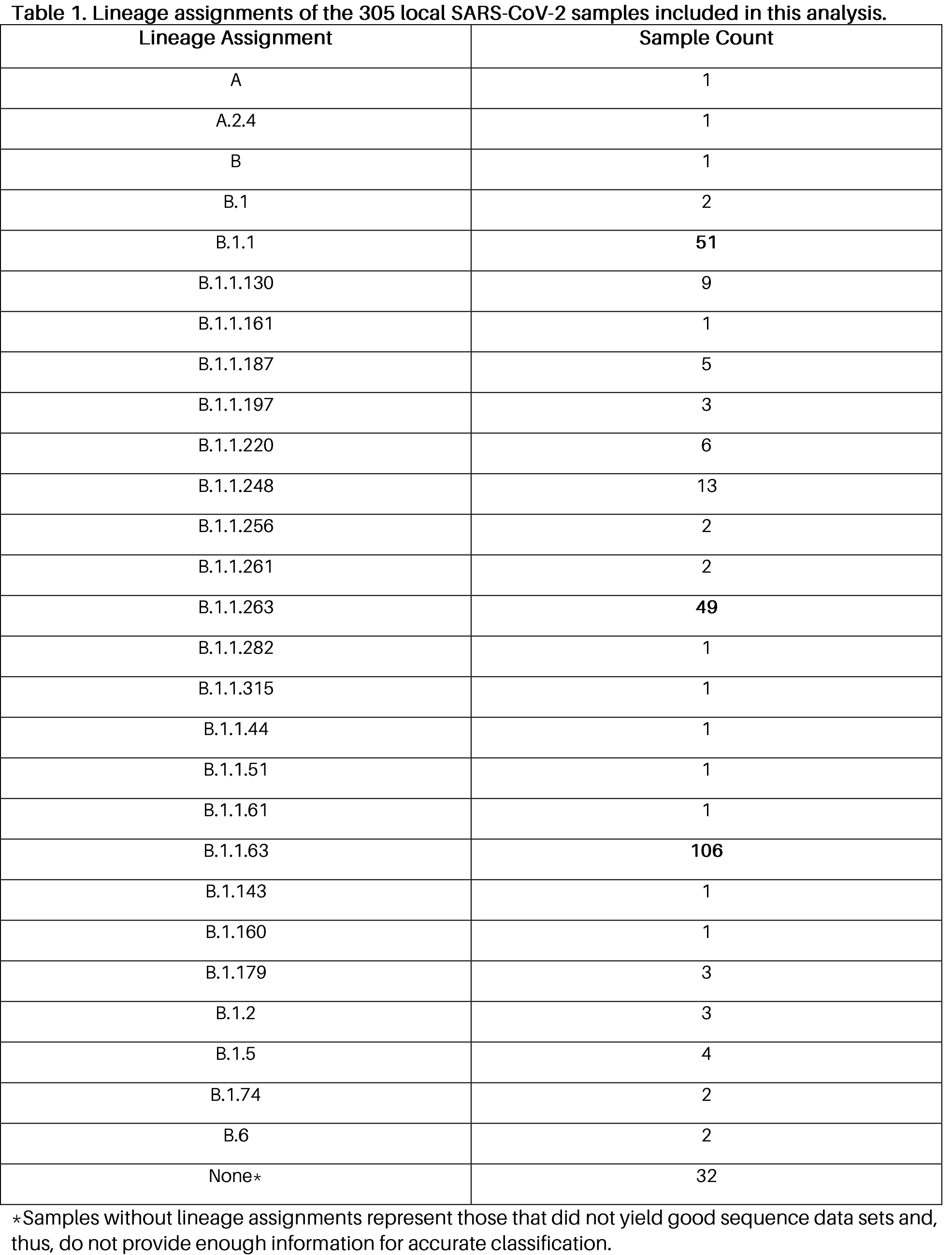
The most frequent spike protein mutations (Figure 1) observed in the local samples are D614G (264 consensus sequences) and S12F (103 consensus sequences), which is similar to our previous observations (PGC SARS-CoV-2 Bulletin No. 4). These mutations have already been observed previously, but their effects are not yet fully characterized apart from previous reports that the D614G mutation may increase infection rate. Note, however, that for the samples that did not yield good data sets and were not assigned lineages, we cannot say for certain whether or not the defining mutations for the UK and South African variants are present.
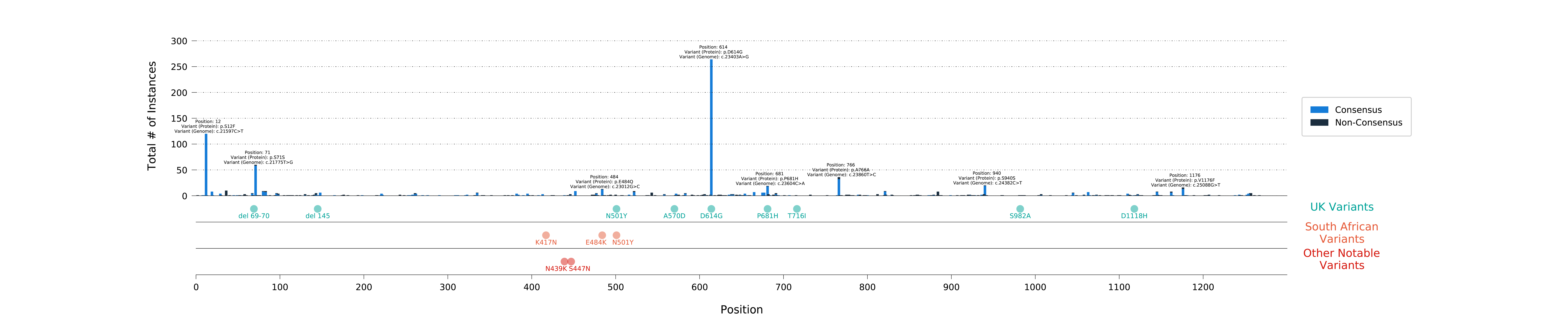
Figure 1. Spike protein mutations found in local SARS-CoV-2 isolates. A total of 305 Philippine SARS-CoV-2 isolate genomes were sequenced and analyzed. The plot shows the number of samples wherein a particular spike protein mutation has been observed. Consensus mutations (blue bar) refer to genetic changes found at high frequencies and are thereby reflected in the consensus sequences, while non-consensus mutations (black bar) are those observed at relatively lower frequencies within the samples. Typically, high frequency mutations are more likely to be dominant and have greater chances of spreading. The defining mutations of the UK variant, South African variant, and other variants of interest are also shown for comparison.
Based on the analysis of 305 samples, we still don’t have evidence that the UK and South African variants of SARS-CoV-2 have entered the country. However, we need to continue and expand our biosurveillance efforts together with the DOH EB, the RITM and the UP NIH, particularly to cover historical samples, as well as those from areas with recent clusters of infection, inbound travelers from high-risk countries, and hospitalized patients from additional regions in order to monitor changes within the circulating local viral populations.
Reference
PGC SARS-CoV-2 Bulletin No. 4 (2020) Newly reported SARS-CoV-2 variant in the United Kingdom has yet to be observed locally. https://pgc.up.edu.ph/pgc-sars-cov-2-bulletin-no-4/