-
- Through the biosurveillance efforts of the UP Philippine Genome Center (PGC), National Institutes of Health (NIH), and the Department of Health (DOH), a SARS-CoV-2 variant bearing multiple mutations of concern in the spike protein region has been first detected through whole genome sequencing at the UP PGC DNA Sequencing Core Facility and genomically characterized in the Philippines, and was officially designated as Lineage P.3 on March 10, 2021.
- The official designation of this new variant as Lineage P.3 was based on an initial set of 33 sequences deposited by the UP-PGC in the EpiCoVᵀᴹ database of the Global Initiative for Sharing All Influenza Data (GISAID) on March 03, 2021, under accession codes EPI_ISL_1122426 to EPI_ISL_ 1122458.
- A preprint describing in detail the detection and genomic analysis of the first 33 cases of P.3 viruses initially identified in the country has been submitted in medRxiv on March 03, 2021 and publicly posted on March 06, 2021.
- The P.3 cases were initially identified from a next generation sequencing (NGS) run on February 04, 2021 and have since been detected in all subsequent sequencing batches, with currently nearly a hundred cases under this new lineage. The earliest case was collected on January 8, 2021.
- The collection dates of the samples detected to be under this lineage coincide with a sharp increase in SARS-CoV-2 infections in Central Visayas.
- The reported variant carries multiple mutations in the spike protein of the virus that are likely to have functional significance. In particular, the E484K, N501Y, and P681H mutations have been previously associated with known SARS-CoV-2 variants of concern and, together with a three (3) amino acid deletion at spike positions 141 to 143, have been linked to possible increased transmissibility and immune escape in some studies.
- The combination of mutations observed in this variant warrants further studies in order to determine their collective effects on the transmissibility (rate of infection), pathogenicity (severity of the disease), and immunogenicity (impact on vaccine efficacy) of the virus.
- The potential public health implications of the mutations call for strengthened measures to prevent the spread of P.3 to nearby islands of the archipelago and to other countries.
Technical Details
The University of the Philippines – Philippine Genome Center and National Institutes of Health in coordination with the Department of Health and the Philippine Genomic Biosurveillance Network, was able to detect SARS-CoV-2 cases initially classified to be under lineage B.1.1.28, but with the co-occurrence of the E484K and N501Y spike protein mutations. These mutations have been previously associated with SARS-CoV-2 variants of concern (B.1.1.7, B.1.351, P.1) and evidence of their roles in increased transmissibility and viral immune escape has also been reported.1,2,3 This particular variant of SARS-CoV-2 has now been officially designated on March 10, 2021 as lineage P.3 – an alias of B.1.1.28.3, alluding to their shared ancestry with the B.1.1.28 viruses that also gave rise to the P.1 variant of concern.
A preprint describing in detail the detection and genomic analysis of the first 33 cases of P.3 viruses initially identified in the country is now publicly available through the medRxiv server.4 The figures shown in this report are derived from the said preprint. Additionally, a preprint of a follow up study on the possible structural implications of some of the mutations observed in this variant is now posted in bioRxiv.5
The first of such cases was identified from a sequencing run last February 04, 2021, and has been detected in all of the subsequent sequencing batches since then. To date, a total of 98 sequenced samples can be classified under this lineage, majority of which were collected from Central Visayas. Interestingly, the recorded collection dates of these samples coincide with a sharp increase in the number of SARS-CoV-2 infections registered in the said region. The geographic distribution of the first 33 detected P.3 cases (unofficially designated as PHL-B.1.1.28) is shown in Figure 1A.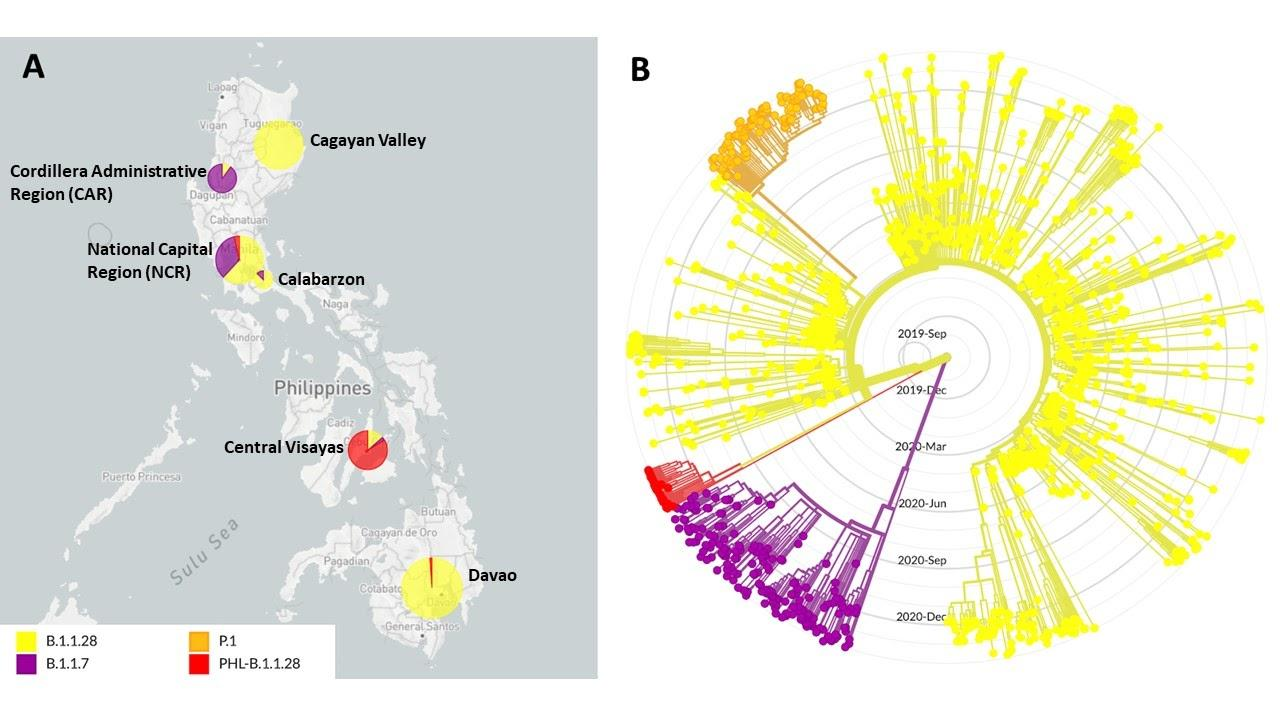
Figure 1. Geographic distribution and phylogenetic clustering of an emergent SARS-CoV-2 lineage in the Philippines designated as PHL-B.1.1.28. (A) Geographic distribution of viruses under lineages B.1.1.28 (Yellow), B.1.1.7 (Purple), P.1 (Orange) and the emergent variant PHL-B.1.1.28 (Red, now designated as P.3) in the Philippines. Most of the samples detected to have the signature mutations of PHL-B.1.1.28 (P.3) were from the Central Visayas region. (B) Phylogenetic tree of B.1.1.28, B.1.1.7, P.1, and PHL-B.1.1.28 (P.3) sequences showing that the emergent variant forms a monophyletic clade separate from other lineages included in the analysis. The B.1.1.7 and B.1.1.28 clades include samples collected both globally and in the Philippines.
Phylogenetic reconstruction of the initial 33 P.3 cases detected in the country, with representative samples from the B.1.1.7 and B.1.1.28 lineages, including the P.1 lineage which is also derived from B.1.1.28, indicate that they represent an emergent lineage within the global phylogenetic tree (Figure 1B). The phylogeny reveals that these samples cluster into a well-defined monophyletic clade (red) that diverges from the B.1.1.28 lineage (yellow). Notably, the tree topology shows that this clade does not derive from the P.1 lineage, indicating that the E484K and N501Y mutations were not inherited from P.1 viruses but were most likely independently acquired. This acquisition may have occurred during a chronic or persistent infection such as in immunocompromised patient(s),6 as well as during continued, uninterrupted transmission of the virus that allows them to evolve further from one host to the other.

Figure 2. Mutation profile of 33 Philippine SARS-CoV-2 samples comprising an emergent viral lineage. A total of 14 amino acid replacements were observed in all samples (labeled in red), including seven spike protein mutations. Among the spike protein mutations, four have been previously associated with lineages of concern (i.e., E484K, N501Y, D614G, and P681H) while three additional replacements were observed towards the C-terminal region of the protein (i.e., E1092K, H1101Y, and V1176F). Interestingly, a single amino acid replacement at the N-terminus of ORF8 (i.e., K2Q) was also found in all samples. Three other mutations were seen in 32 of the 33 samples (labeled in green) including a three-amino acid deletion at the spike protein positions 141 to 143. Lastly, five synonymous mutations (labeled in gray) were also detected in all of the cases. For comparison, the defining mutations for the P.1, B.1.1.7, and B.1.351 lineages of concern are marked below in red, blue, and green boxes, respectively. None of the samples exhibit mutation profiles similar to any of the three known lineages of concern.
A close examination of the mutation profile of the P.3 viruses revealed that apart from E484K and N501Y, they also share other spike protein mutations that are likely to have functional significance (Figure 2). This includes the P681H mutation, also found in lineage B.1.1.7 viruses, as well as a three-amino acid deletion at positions 141 to 143 (LGV141_143del). Deletions in this region of the spike protein has been implicated in possible immune escape mechanisms.7 Thus, in combination with other radical changes observed in the spike protein and other regions of the genome, the effect of these mutations on the transmissibility, pathogenicity, and immunogenicity of the virus warrants further experimental (laboratory) and epidemiological (distribution and frequency) investigation. It also merits enhanced measures to contain the spread of the variant to other islands in the archipelago and to other countries.
References
1 Collier, D., De Marco, A., Ferreira, I., Meng, B., Datir, R., Walls, A. C., … & CITIID-NIHR BioResource COVID-19 Collaboration. (2021). SARS-CoV-2 B.1.1.7 sensitivity to mRNA vaccine-elicited, convalescent and monoclonal antibodies. medRxiv. doi: https://doi.org/10.1101/2021.01.19.21249840
2 Leung, K., Shum, M. H., Leung, G. M., Lam, T. T., & Wu, J. T. (2021). Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance, 26(1), 2002106.
3 Luan, B., Wang, H., & Huynh, T. (2021). Molecular Mechanism of the N501Y Mutation for Enhanced Binding between SARS-CoV-2’s Spike Protein and Human ACE2 Receptor. bioRxiv, 2021-01. doi: https://doi.org/10.1101/2021.01.04.425316
4 Tablizo, F. A., Kim, K. M., Lapid, C. M., Castro, M. J. R., Yangzon, M. S. L., Maralit, B. A., … & Saloma, C. P. (2021). Genome sequencing and analysis of an emergent SARS-CoV-2 variant characterized by multiple spike protein mutations detected from the Central Visayas Region of the Philippines. medRxiv. doi: https://doi.org/10.1101/2021.03.03.21252812
5 Bascos, N. A. D., Mirano-Bascos, D., & Saloma, C. P. (2021). Structural Analysis of Spike Protein Mutations in an Emergent SARS-CoV-2 Variant from the Philippines. bioRxiv. https://doi.org/10.1101/2021.03.06.434059
6 Kemp, S. A., Collier, D. A., Datir, R. P., Ferreira, I. A. T. M., Gayed, S., Jahun, A., … & Gupta, R. K. (2021). SARS-CoV-2 evolution during treatment of chronic infection. Nature. https://doi.org/10.1038/s41586-021-03291-y
7 McCarthy, K. R., Rennick, L. J., Nambuli, S., Robinson-McCarthy, L. R., Bain, W. G., Haidar, G., & Duprex, W. P. (2021) Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science. eabf6950. DOI: 10.1126/science.abf6950.